
LATER: Installing a Dimmer Switch for Pain
A new way to turn down pain

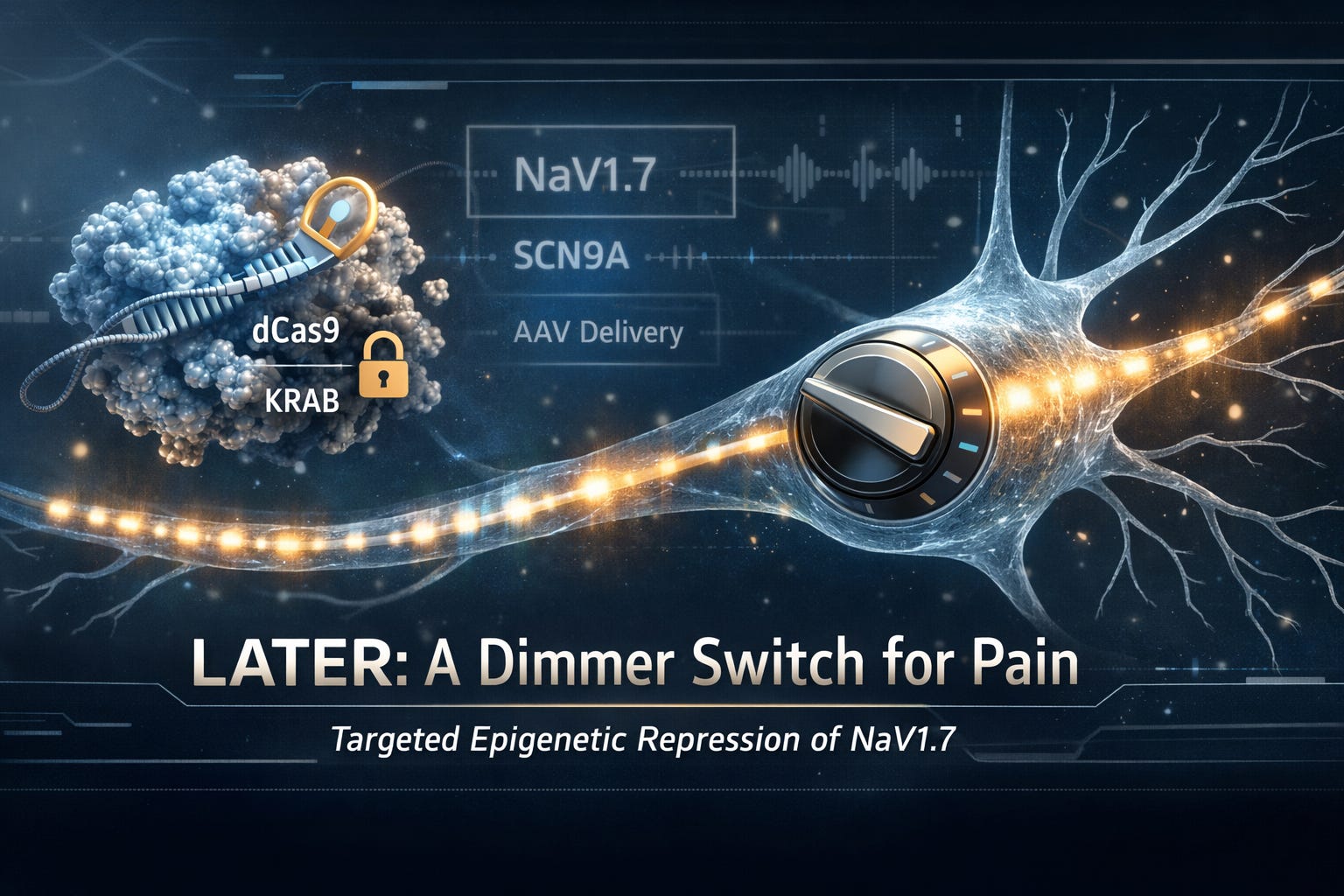
If you have ever had electrical work done, you know the blunt approach: cut the wire, flip the breaker, and the signal stops. Pain medicine has often worked the same way, either by broadly blocking nerve activity (think local anesthetics) or by dulling the brain’s perception of pain (think opioids). Both can work, but both come with tradeoffs, numbness, sedation, tolerance, addiction risk, and lots of biological “collateral damage.”
Now imagine a different approach: instead of cutting wires, you install a dimmer switch. The circuit still exists. The system still functions. You just turn the signal down, precisely, at its source.
That is the promise of LATER, short for Long-lasting Analgesia via Targeted Epigenetic Repression. In preclinical work, LATER uses programmable gene-targeting tools to temporarily silence a key pain gene, SCN9A, which encodes the NaV1.7 sodium channel, without cutting DNA. The goal is long-term pain relief after a single treatment, with a risk profile that looks more like “gene regulation” than “permanent gene editing.”
The science, explained simply
Pain is an electrical language
Pain begins in specialized nerve cells called nociceptors, your body’s early warning system for tissue damage. When they detect injury or inflammation, they generate electrical impulses that travel to the spinal cord and then the brain.
Those electrical impulses rely on tiny molecular gates in the nerve membrane called ion channels. Think of them as turnstiles for charged particles. When certain channels open, ions flow, voltage changes, and the neuron fires a signal.
Meet NaV1.7, a crucial “gain knob” in pain-sensing neurons
NaV1.7 is a voltage-gated sodium channel heavily involved in pain signaling in peripheral sensory neurons. It is encoded by the gene SCN9A. One reason NaV1.7 has become a “holy grail” pain target is that human genetics has already run the experiment for us:
People with loss-of-function SCN9A mutations can have congenital insensitivity to pain, meaning they do not feel pain despite otherwise typical development.
People with certain gain-of-function SCN9A mutations can experience severe pain syndromes, such as primary erythromelalgia (burning pain with redness and warmth, often triggered by heat or exercise).
Other SCN9A variants cause syndromes like paroxysmal extreme pain disorder, featuring intense episodic pain.
This is why scientists keep coming back to NaV1.7: dialing it down should reduce pain, and nature suggests you might do that without shutting down the nervous system entirely.
CRISPR, but not the scissors version
CRISPR as a GPS, not a blade
Many people hear “CRISPR” and think “gene editing,” meaning cutting DNA and changing the genetic code. That is one use. But CRISPR can also be repurposed as something more like a programmable GPS-enabled clamp.
CRISPR systems use a small piece of RNA (a guide RNA) to find a matching DNA sequence. The protein Cas9 is the “hands” that follow the guide. In classic editing, Cas9 cuts.
In LATER, researchers use a modified form called dead Cas9, written dCas9. It is “dead” only in the sense that it cannot cut DNA. It can still bind very precisely to a target location.
So instead of scissors, you now have a programmable docking device that parks on a gene like SCN9A exactly where you want it.
KRAB: turning binding into silencing
A programmable “off switch” for a pain gene
Binding alone does not silence a gene. To actually turn down SCN9A, LATER couples dCas9 to a powerful repression domain called KRAB (Krüppel-associated box).
Here is the intuition:
dCas9 is the address label and the delivery truck that brings you to the right house (the SCN9A gene).
KRAB is the locksmith who shows up and quietly changes the locks so the gene’s “readers” cannot get in.
At the molecular level, KRAB recruits co-repressor machinery (often described through the KAP1/TRIM28 hub) and associated enzymes that lay down repressive chromatin marks such as histone H3 lysine 9 methylation, helping attract proteins like HP1 and promoting a compact, silenced chromatin state.
The practical takeaway: you can target KRAB-dCas9 to SCN9A and reduce NaV1.7 production without changing the underlying DNA letters.
Delivery: how do you get it into the body?
AAV is the delivery vehicle
A brilliant molecular tool is useless if it cannot reach the right cells. LATER’s preclinical strategy uses AAV, short for adeno-associated virus, as a delivery vehicle.
AAV is not used because it causes disease. It is used because it is a highly evolved nanoscale courier: it can enter cells and deliver genetic instructions. In gene therapy, engineered AAV vectors have become a workhorse because they can drive long-term expression in certain tissues.
Why AAV is a strong fit for neurons
Neurons are mostly non-dividing cells, and AAV-delivered DNA often persists in the nucleus largely as episomal DNA (separate from chromosomes). In non-dividing cells, that persistence can translate into long-lived production of the therapeutic payload.
For pain, a key anatomical target is the dorsal root ganglion (DRG), clusters of sensory neuron cell bodies that act like relay hubs for signals entering the spinal cord. In the foundational LATER mouse work, AAV was delivered intrathecally (into the cerebrospinal fluid around the spinal cord), a route known to access DRG neurons.
What makes it epigenetic?
Closing the book, not rewriting the text
A useful way to think about DNA is as a library. Every cell has the same books (genes), but not every cell reads every book. The difference is controlled by chromatin, the packaging of DNA around histone proteins.
Open, accessible chromatin is like a book laid open on a desk.
Repressed chromatin is like a book shut tight and taped closed.
Epigenetics is about changing how readable a gene is, not changing the gene’s spelling. LATER aims to silence SCN9A by nudging it into a more heterochromatin-like, compact state via KRAB-recruited machinery.
That distinction matters because it reframes the therapy from “editing the genome” to “training gene expression.”
How long does it last?
Months of relief from a single dose in mice
In the key mouse study that coined “pain LATER,” researchers delivered AAV carrying either a KRAB-dCas9 system or a zinc finger-KRAB repressor system targeting NaV1.7. They tested multiple pain models including inflammatory and neuropathic paradigms, and reported reduced pain hypersensitivity with no obvious motor deficits.
Most strikingly, they evaluated durability in an inflammatory pain model and reported efficacy at 42, 84, and 308 days after a single intrathecal AAV injection, which is on the order of many months to nearly a year in a mouse lifespan.
Epigenetic memory, the cell “remembers” the dimmer setting
Why might repression persist so long? Part of the answer is that chromatin states can exhibit epigenetic memory. Once a locus is pushed into a repressed configuration, the molecular marks and the proteins that recognize them can reinforce that state over time, even as the cell’s molecular components turn over. KRAB-associated repression has also been shown to support long-range repression through heterochromatin-like spreading in model systems.
Safety value of reversibility
Classic CRISPR editing can create permanent DNA changes. LATER is conceptually different: it aims for durable regulation rather than permanent rewriting. CRISPR interference-style approaches are often described as reversible in principle because if the repressor is removed, transcription can return.
In practice, long-lived delivery (like AAV in neurons) makes “turning it off” a real design challenge, but the underlying biology still offers a crucial safety concept: no intentional DNA cutting is required to get long-lasting effect.
Who’s developing this?
Navega Therapeutics and an epigenetic approach to chronic pain
Navega Therapeutics is a San Diego biotech built around the idea that chronic pain can be treated by precisely turning down SCN9A/NaV1.7 expression using targeted epigenetic regulation. The company has described its lead program NT-Z001 as an epigenetic therapy targeting SCN9A (NaV1.7) for chronic pain conditions including rare and severe disorders such as primary erythromelalgia and small fiber neuropathy.
The company has also publicly discussed advancing toward the clinic with support for IND-enabling toxicology and other preclinical development work, including through SBIR funding aimed at moving toward IND-enabling studies and eventual clinical translation (with an emphasis on non-opioid pain relief, including cancer-related pain settings).
2026 status: late preclinical, scale-up, and first-in-human planning
Based on company announcements and conference materials circulating into 2026, Navega has described NT-Z001 as having completed key safety work in mice and a pilot nonhuman primate study, with IND-enabling studies in their final stages, alongside manufacturing and scale-up steps to support early clinical development.
This is the translational leap that matters: taking a clever gene control trick from animal models and turning it into a product that can be manufactured reproducibly, dosed safely, and tested in humans.
Comparison with competing technologies: RNA editing
RNA editing: rewriting the message, not the blueprint
If LATER targets DNA-level gene expression, RNA editing targets the messenger RNA that carries instructions from DNA to the ribosome, the protein-making machinery.
One family of approaches uses enzymes like ADAR (adenosine deaminases acting on RNA) or engineered systems that recruit editing activity to specific RNAs. Another well-known strategy uses RNA-targeting CRISPR proteins (like Cas13) fused to editing domains to change individual bases on RNA.
In the pain world, an NIH-funded consortium has described site-directed RNA editing of Nav1.7 as a potential analgesic strategy, conceptually “intercepting and editing” the Nav1.7 message before the channel is made.
Why this matters: a post-opioid roadmap for chronic pain
Chronic pain is not just a symptom, it is often a long-term disease state of the nervous system. Many current therapies either mask it or broadly suppress neural activity.
LATER represents a different thesis: pain can be treated by reprogramming gene expression in the peripheral nervous system, selectively reducing a biologically validated amplifier of pain signaling (NaV1.7) while leaving the rest of the wiring intact.
If this general strategy translates safely to humans, the real-world impact could be enormous:
fewer patients funneled toward addictive drugs,
more targeted relief for severe neuropathic pain,
and the possibility that a single treatment could buy months of meaningful function.
It is not science fiction. It is gene regulation, delivered like a therapy.
The frontier question now is not whether we can build the dimmer switch. We can. The question is whether we can install it safely, tune it precisely, and prove in humans that turning down one channel can turn down suffering.
References (primary literature and primary scientific sources)
Moreno AM, Aleman F, Catroli GF, et al., 2021, Long-lasting analgesia via targeted in situ repression of NaV1.7 in mice, Science Translational Medicine.
Cox JJ, Reimann F, Nicholas AK, et al., 2006, An SCN9A channelopathy causes congenital inability to experience pain, Nature.
Yang Y, Wang Y, Li S, et al., 2004, Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia, Journal of Medical Genetics.
Fertleman CR, Baker MD, Parker KA, et al., 2006, SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes, Neuron.
Qi LS, Larson MH, Gilbert LA, et al., 2013, Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression, Cell.
Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ, 2002, SETDB1: a novel KAP1-associated histone H3 lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins, Genes and Development.
Groner AC, Meylan S, Ciuffi A, et al., 2010, KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading, PLoS Genetics.
Cox DBT, Gootenberg JS, Abudayyeh OO, et al., 2017, RNA editing with CRISPR-Cas13, Science.
Katrekar D, Chen G, Meluzzi D, et al., 2019, In vivo RNA editing of point mutations via RNA-guided adenosine deaminases, Nature Methods.
Katrekar D, Yen J, Xiang Y, et al., 2022, Efficient in vitro and in vivo RNA editing via recruitment of endogenous ADARs using circular guide RNAs, Nature Biotechnology.
Gordon EA, et al., 2025, Selective targeting of voltage-gated sodium channels to treat pain (includes site-directed RNA editing of Nav1.7 concept), PAIN (conference abstract).
Absolutley fascinating breakdown of the LATER approach. The framing of dCas9-KRAB as a 'dimmer switch' rather than scissors finally gave me a clear mental model for understanding epigenetic repression vs gene editing. What excites me most is the duration of effect in the mouse studies, months of relief from a single dose seems almost too good to be true but the epigentic memory mechanism explains it well. Really hoping Navega's clinical trials move forward smoothly.